Información útil para el médico sobre la Esclerosis Múltiple
.
Su prevalencia es variable, siendo más frecuente en regiones que se encuentran en latitudes mayores a los 45° como Europa, el norte de E.U.A, Canadá o Australia alcanzando rangos de prevalencia de hasta 50 a 100 pacientes por 100, 000 habitantes. Mientras que en México la prevalencia varia de cuerdo a la región del país entre 5 hasta 18 pacientes por cien mil habitantes. Los síntomas neurológicos más frecuentes son las alteraciones sensitivas, visuales, motoras, de la coordinación y del equilibrio, por mencionar algunas.
.
Existen varios tipos de Esclerosis Múltiple entre ellas está la EM remitente recurrente (EMRR), EM secundaria progresiva (EMSP) y EM Primaria progresiva (EMPP), definidas de esta forma de acuerdo a la aparición y evolución de sus síntomas. El diagnóstico de hace por medio de los criterios de Mc Donald del 2017 basados en datos clínicos e imagenológicos provenientes de la RM de encéfalo y médula espinal, aunque existen otros estudios que se realizan para descartar la presencia de enfermedades que presenten los mismos síntomas o para identificar el pronóstico o grado de daño de las estructuras del sistema nervioso central (SNC). En la actualidad, existe múltiples fármacos para evitar la aparición de brotes o progresión, conocidos como terapia modificadora de la enfermedad (TME); además de medicamentos para tratar los síntomas específicos.
La EM es considerada la principal causa de discapacidad no ocasionada por traumatismo en jóvenes, razón por la cual es muy importante identificar su frecuencia, distribución y factores de riesgo y pronóstico. La prevalencia de EM ha incrementado desde la década de los 50´s; de acuerdo con la Federación Internacional de EM, en 2013 se estimó una prevalencia mundial de la enfermedad de 2.3 millones de personas. (2) Haciendo un mapeo de la enfermedad, se ha identificado un patrón geográfico definido de la frecuencia, ubicando mayor cantidad de pacientes en aquellos países que se encuentran en latitudes mayores a 45° al sur y al norte, como Europa, el norte de U.S.A, Canadá, Australia o Argentina, con prevalencias altas de entre 50 a 100 pacientes por 100 000 habitantes. Esto es debido a que la EM afecta principalmente a personas caucásicas o descendientes de europeos, resultando raro o ausente su diagnóstico en asiáticos, mestizos, afroamericanos o amerindios (1).
Una publicación reciente mostro la variabilidad de la prevalencia en Latinoamérica la cual se calcula entre 5 a 15 pacientes por 100 000 habitantes aproximadamente, teniendo a Argentina posicionada como el país latinoamericano con la mayor prevalencia ( 38.2/105) y Ecuador con la menor (0.75/105); en tanto que en México es de 7-8 pacientes por 100 000 habitantes en general, con una región en el norte de hasta 30 pacientes por cien mil habitantes. (3)
Las características epidemiológicas varían de acuerdo al tipo de EM; en el caso de EMRR, la más frecuente hasta en un 85%, afecta con mayor frecuencia a mujeres (3:1 M:H) de 20 a 35 años de edad; en tanto que la EMPP afecta más a hombres después de los 40 años.(1,4)
El daño ocasionado en la mielina del SNC es resultado de un proceso inflamatorio agudo y crónico mediado por un complejo mecanismo fisiopatológico en el que están involucrados el sistema inmune, la glia y las neuronas, tal y como lo menciona Reich y cols. (4) La característica patológica distintiva de la EM es la presencia de lesiones inflamatorias perivenulares que terminarán en las placas desmielinizantes y pérdida de la integridad de la barrera hematoencefálica, tanto en la sustancia blanca como en la sustancia gris del encéfalo, médula espinal y nervio óptico. (5,6) Los eventos necesarios para llegar a este resultado son desencadenados por la actividad de Linfocitos T y B activados erróneamente probablemente por autoantígenos, aunque hasta el momento ninguno ha sido confirmado como el responsable absoluto de la enfermedad. Hay dos teorías de por qué se genera esta respuesta inmune de intolerancia: una de ellas es llamada “intrínseca” en la cual el evento inicial toma lugar en el SNC donde se cree hay una liberación de antígenos que después salen a la periferia; y la otra llamada “extrínseca” donde todo inicia a nivel sistémico tal vez ocasionado por una infección viral. (5)
Un evento agudo o brote de EM es secundario a la presencia de una lesión desmielinizante inflamatoria aguda o activa, la cual está constituida por un infiltrado de linfocitos reactivos, especialmente Linfocitos T CD8+ y B CD20+, pero también algunos Linfocitos CD4+, microglía activada, macrófagos espumosos y astrocitos. Por otro lado, las lesiones crónicas presentan bordes bien circunscritos, son hipocelulares y tienen datos francos de desmielinización, reducción de la densidad axonal, gliosis y activación de la microglía únicamente en los bordes la placa, escasa cantidad de linfocitos y no cuentan con la presencia de macrófagos. (1)
En un inicio el axón recubierto por el oligodendrocito sufre un daño leve o se mantiene intacto, pero a lo largo del tiempo este daño incrementa e incluso llega a provocar disfunción irreversible neuronal. El resultado de todo este complejo proceso inflamatorio crónico y agudo es la desmielinización, pérdida axonal y gliosis del tejido nervioso impidiendo su función. (5)
La fisiopatología inmunológica tradicionalmente aceptada involucraba un daño mediado principalmente por linfocitos T, que incluía una disfunción de los linfocitos T reguladores (Treg). De tal forma que se proponía que el linfocito T erróneamente activado ingresaba al SNC a través de la barrera hematoencefalica dañada por metaloproteinasa, entre otras sustancias, y una vez adentro el linfocito sufría una expansión clonal, para después diferenciarse en Linfocitos T cooperadores o helper tipo 1 o tipo 2 (LTH) , provocando la activación de macrófagos , linfocitos citotóxicos y células B, con lo que se genera un daño directo a la mielina del oligodendrocito a través de citotoxicidad y autoanticuerpos, (7) apoyado por la secreción de citocinas, quimiocina y especies reactivas de oxígeno, las cuales promueven el daño local y la neurodegeneración. Una de las moléculas proinflamatorias más importante secretadas por los Linfocitos T helper 17 (LTH 17) y CD8+ es la interleucina 17(IL17), pero también existen otras sustancias trascendentes como el interferón gamma (IFNγ) secretado por LT CD4+, y el factor estimulante de colonias de granulocitos y macrófagos (GM-CSF), expresado por LT CD4+ y CD8+.
Actualmente esta visión está siendo renovada debido al papel fundamental que han demostrado tener los linfocitos B. Basados en la elevada producción de anticuerpos y la presencia de bandas oligoclonales en líquido cefalorraquídeo (LCR), se propone que los Linfocitos B CD20+ tienen un papel preponderante en la fisiopatología de la EM. Las células B en pacientes con EM tienden a producir citocinas proinflamatorias, pero también a limitar la secreción de interleucinas antiinflamatorias como la IL-10. En este proceso también está involucrada una disfunción de células B reguladoras, capaces de modular la actividad y producción de citocinas y antocuerpos. Este desequilibrio en el funcionamiento del Linfocito B promueve una respuesta anómala de los linfocitos TH1 y TH17, generando también citocinas proinflamatorias y daño celular al tejido nervioso, especialmente a la mielina.(1,7)
Existen 4 tipos de EM de acuerdo su curso clínico: EM remitente recurrente (EMRR), EM secundaria progresiva (EMSP), EM primaria progresiva (EMPP) y EM progresiva remitente (EMPR), aunque en la actualidad las variantes aceptas son las primeras tres. De todas , la más frecuente es la EMRR con una frecuencia del 85%; su característica principal es la presencia de episodios llamados brotes o recaídas en la cual aparecen síntomas neurológicos que cuestión de horas cuya condición es que duren más de 24 horas, para posteriormente incrementar en intensidad hasta alcanzar su máximo pico y después disminuir en días o semanas, aproximadamente 3 o 4; para considerarlo un brote, es importante que la sintomatología no esté acompañada de fiebre, un proceso infeccioso, o las características clínicas de encefalitis como alteraciones del estado de despierto o crisis epilépticas, por ejemplo. La presencia de los brotes es totalmente impredecible tanto en tiempo como en el tipo de sintomatología, pero se ha establecido un promedio de frecuencia de brotes de 1,1 al año; desafortunadamente, estos eventos pueden dejar secuelas irreversibles en ocasiones. Un 2 a 3 % de los pacientes con EMRR después de 15 a 20 años de evolución, convierten a EMSP, la cual se caracteriza por aparición de sintomatología neurológica irreversible y discapacitante de progresión lenta y de difícil percepción en comparación con los brotes.
La EMPP representa el 10 al 15% de la EM y está caracterizada por síntomas progresivos desde el inicio de la enfermedad dando como resultado una discapacidad irreversible en un año y no presenta brotes.
Un concepto de gran interés es el Síndrome Clínico aislado o CIS por sus siglas en inglés, el cual se refiere a paciente que presentan su primer brote por manifestaciones clínicas pero que no cumplen con los criterios radiológicos para complementar el diagnóstico. Este síndrome a su vez puede clasificarse como CIS activo o no activo, dependiendo de la presencia de recaídas o lesiones detectadas en RM; de acuerdo a la presencia de actividad se toma la decisión de iniciar TME.(1)
También existe el Síndrome radiológico aislado, donde el paciente esta asintomático, pero se han encontrado lesiones desmielinizantes en RM, pero aun sin cumplir criterios para EM.
La presencia de síntomas en los pacientes con EM es muy variable, ya que esta depende de la localización de la lesión activa en el SNC. Dentro de los síntomas más frecuentes al inicio de la enfermedad están los de tipo sensitivo (hasta en un 43%), motores (30-40%), vértigo, disminución de agudeza visual o defecto campimétrico, alteraciones de la coordinación o equilibrio o problemas en el control de esfínteres. La neuritis óptica suele aparecer en el primer episodio hasta en un 25% de los pacientes y hasta el 70% de los pacientes la sufrirá durante algún momento de enfermedad.
Otros síntomas que se pueden presentar son: deterioro cognitivo, fatiga, alteraciones del estado de ánimo (depresión como la más frecuente), trastornos del sueño (insomnio, apnea obstructiva del sueño o síndrome de piernas inquietas) y dolor, que no es frecuente, pero puede manifestarse como neuralgia del trigémino, espasticidad, lumbalgia o dolor visceral.
El diagnostico está basado en criterios de Mc Donald 2017, que toman en cuenta la clínica por la presencia de brotes, pero también se apoya en estudios de imagen como la RM. (8)
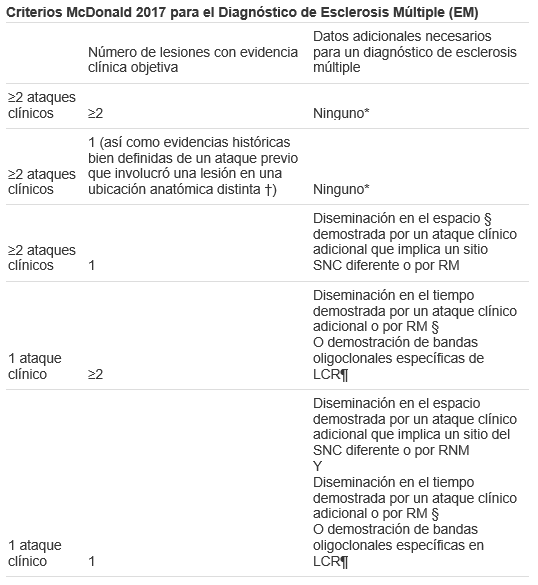
* No se requieren pruebas adicionales para demostrar la diseminación en espacio y tiempo. Sin embargo, a menos que la RM no sea posible, se debe obtener una RM cerebral en todos los pacientes en quienes se está considerando el diagnóstico de esclerosis múltiple. Además, se debe considerar el examen de RM o LCR de la médula espinal en pacientes con evidencia clínica y de RM insuficiente que apoyan la esclerosis múltiple, con una presentación que no sea un síndrome clínico típico aislado, o con características atípicas. Si las imágenes u otras pruebas (por ejemplo, LCR) son negativas, se debe tener precaución antes de hacer un diagnóstico de esclerosis múltiple, y se deben considerar diagnósticos alternativos.
El diagnóstico clínico basado en hallazgos clínicos objetivos de dos ataques es más seguro. La evidencia histórica razonable de un ataque pasado, en ausencia de hallazgos neurológicos objetivos documentados, puede incluir eventos históricos con síntomas y características de evolución para un ataque desmielinizante inflamatorio previo; al menos un ataque, sin embargo, debe ser respaldado por hallazgos objetivos. En ausencia de evidencia objetiva residual, se necesita precaución.
Los criterios de RM para diseminación en espacio y tiempo son:
- La diseminación en el espacio se puede demostrar por una o más lesiones hiperintensas en T2 que son características de la esclerosis múltiple en dos o más de cuatro áreas del SNC: regiones periventriculares, cortical o yuxtacortical e infratentorial, y la médula espinal
- La diseminación en el tiempo se puede demostrar mediante la presencia simultánea de lesiones reforzadas con gadolinio y no reforzadas* en cualquier momento o mediante una nueva lesión hiperintensa en T2 o reforzada con gadolinio en la RM de seguimiento, con referencia a una exploración basal, independientemente del momento de la RM de referencia
La presencia de bandas oligoclonales específicas de LCR no demuestra la diseminación a tiempo per se, pero puede sustituir el requisito de demostración de esta medida.
El objetivo del médico que atiende a pacientes con EMRR es lograr un estado de ausencia de actividad de la enfermedad. Para esto se ha creado NEDA (NO evidencia de actividad de la enfermedad) el cual es una serie de parámetros que establecen las metas para alcanzar un control óptimo de la enfermedad. Hasta el momento, NEDA 3 es el más accesible y utilizado por los neurólogos, ya que consta de la ausencia de recaídas y progresión de la enfermedad clínica (NEDA 1-2) y falta de actividad en RM (NEDA-3). El número de NEDA continua en crecimiento, siendo NEDA 4 un control de la atrofia cerebral y NEDA 5 la medición de neurofilamento del LCR, lo cual muchas veces no es posible en todas las unidades hospitalarias. (9,10)
Para proporcionar un abordaje terapéutico completo en el paciente con EM se debe tomar en cuenta un tratamiento multidisciplinario que incluya la administración de medicamentos para tratar el brote en agudo, evitar la aparición de brotes y discapacidad (llamada tratamiento modificador de la enfermedad) y tratamiento sintomático.
El tratamiento de la EM se divide en tres rubros principales:
- Tratamiento en caso de Brote :
- Metilprednisolona.
- Tratamiento modificador del curso de la enfermedad (TME):
- Inmunosupresores.
- Inmunomoduladores.
- Tratamiento sintomático-multidisciplinario:
- Medicamentos específicos.
- Rehabilitación.
- Atención psicológica, psiquiátrica y de otras especialidades.
Tratamiento del episodio agudo o brote
El tratamiento con corticoesteroides es el indicado para brote o recaída por su potenet efecto antinflamatorio. El esquema de aplicación es metilprednisolona 1g IV cada 24 horas por 3 a 5 días de acuerdo al territorio neurológico afectado y a la severidad del brote. En México no se cuenta con metilprednisolona vía oral.
Estas drogas están asociadas con un funcionamiento más rápido recuperación y protección contra la ocurrencia de déficits más graves en las primeras semanas después del tratamiento pero tienen beneficios poco claros a largo plazo.
El uso de plasmaféresis o inmunoglobulinas debe ser considerado en aquellos casos en los que el brote se presenta en forma atípica, fulminante y que no responde a tratamiento esteroideo convencional.
Tratamiento modificador de la enfermedad
EMRR
El TME tiene como objetivo reducir la actividad inflamatoria, modular el sistema inmunológico y con esto evitar la presencia de brotes. En la actualidad contamos con una cantidad aceptable de TME principalmente para EMRR.
Existen criterios para iniciar el TME en EMRR, descritos a continuación:
- Presencia de al menos 2 brotes en los últimos 3 años sin incluir brotes sensitivos subjetivos y tener una puntuación en el EDSS entre 0-5.5.
Actualmente existen diferentes tipos de TME y su elección depende de la eficacia y seguridad del medicamento, así como de las características del paciente. Para realizar la elección existe un sistema de escalada y otro alterno de indicación directa o inducción.
El sistema de escalada propone iniciar con medicamento de plataforma o de primera línea que cuentan con una eficacia moderada pero con una aceptable seguridad. Algunos de ellos son: Interferón, acetato de glatiramer, teriflunomida o dimetilfumarato como primera elección de tratamiento. Si los medicamentos de primera elección fallan (más de dos brotes durante al menos 6 meses de administración del medicamento) o los efectos adversos que produce son muy severos o incapacitantes, está indicado cambiar a medicamentos de segunda línea o de escalada con una mayor potencia como natalizumab, fingolimod, alemtuzumab, dimelfumarato, ocrelizumab o cladribina por ejemplo.
Otra estrategia útil para administrar TME es la de inducción, la cual está basada en la administración de medicamentos de alta potencia desde el inicio de la enfermedad para prevenir la acumulación de daño irreversible y discapacidad. Para la mayoría de los autores, la terapia de inducción se refiere a una inmunointervención en un paciente con factores pronósticos negativos como serian: una EM grave con alta actividad (caracterizado por recaídas graves y frecuentes y/ alta carga lesional o lesiones activas importantes en RM) y una acumulación de discapacidad.
Se diagnostica esclerosis múltiple remitente recurrente grave, agresiva o de gran actividad cuanto tiene uno o más de los siguientes criterios:
- EDSS de 4 puntos en los primeros 5 años del inicio de la enfermedad.
- Dos o más ataques clínicos con resolución incompleta dentro de un año.
- Dos o más estudios de RM con nuevas lesiones activas (captantes de contraste) o incremento en el tamaño de las lesiones en T2, a pesar de tratamiento.
Sin respuesta terapéutica con el uso de uno o más fármaco modificador de la enfermedad durante más de un año.(11)
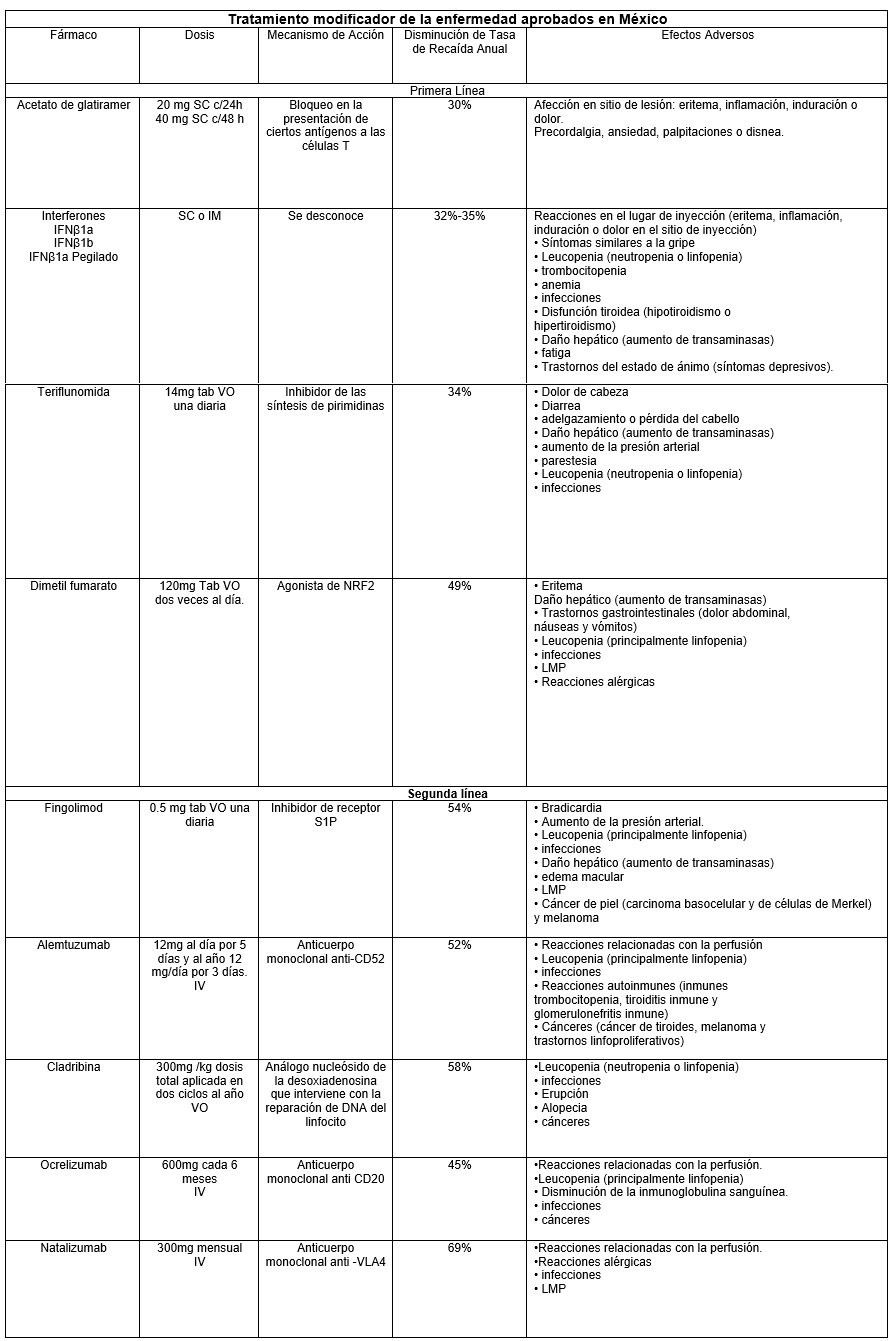
SC: subcutáneo, IM: intramuscular, IV: Intravenoso, VO: vía oral, LMP: Leucoencefalopatía multifocal progresiva.
TME en formas progresivas
EN el caso de EMSP los tratamientos para evitar la progresión son limitados, pero la FDA aprobó el uso de mitoxantrona y recientemente cladribina, específicamente la EMSP con datos de actividad de la enfermedad.
En el caso de EMPP, se cuenta con la aprobación del uso de ocrelixumab, un anticuerpo monoclonal anti CD20, ya que demostró significativamente reducir el riesgo de progresión de la discapacidad en comparación con placebo.
Tratamiento sintomático
Los síntomas que afectan a los pacientes con EM y merman su calidad de vida son dolor, espasticidad, alteraciones de la marcha, pérdida de control de esfínteres o síntomas neuropsiquiátricos.
Los relajantes musculares pueden mejorar la espasticidad en pacientes con EM y la evidencia empírica apoya el uso de baclofeno, dantroleno, tizanidina y toxina botulínica.
En el caso de alteraciones de la marcha, se puede utilizar dalfampridine, un bloqueador de canales de potasio dependiente de voltaje que mejora la transmisión de señales nerviosas en axones desmielinizados.
La rehabilitación física es en muchos casos la piedra angular para el control de espasticidad, paresia, alteraciones de la sensibilidad, re educación de la marcha y falta de coordinación y equilibrio, por lo que esta recomendado su uso a mediano y largo plazo.
En caso de dolor neuropático existen opciones como gabapentina y pregabalina, antidepresivos tricíclicos o inhibidores de la recaptación de serotonina noradrenalina; se deja como segunda opción el uso de opioides.
Para las afecciones en el control de esfínteres se utilizan diferentes medicamentos dependiendo del síntoma predominante ya sea retención urinaria o incontinencia, además de medidas generales o uso de cateterismo intermitente o permanente en caso necesario.




